class: center, middle, inverse, title-slide # A Beginners Guide to Call Somatic Mutation ## Part IV ### Lijia Yu ### 2020/10/05<br/>(updated: 2020-10-05) --- # Outline 0. Review 2. SNP calling: Mutect2 3. VCF format 4. Annotation: Annovar --- # Target Region BED file 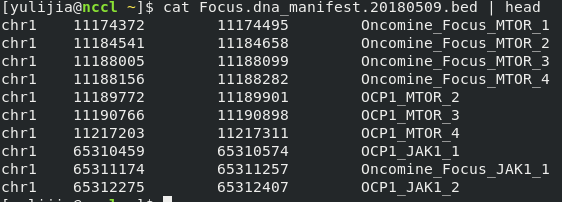 - `chrom`: The name of the chromosome (e.g. chr3, chrY, chr2_random) or scaffold (e.g. scaffold10671). - `chromStart`: The starting position of the feature in the chromosome or scaffold. **The first base in a chromosome is numbered 0.** - `chromEnd`: The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromStart=0, chromEnd=100, and span the bases numbered 0-99. .footnote[ [1][BED format](https://genome.ucsc.edu/FAQ/FAQformat.html#format1) ] --- # Workflow 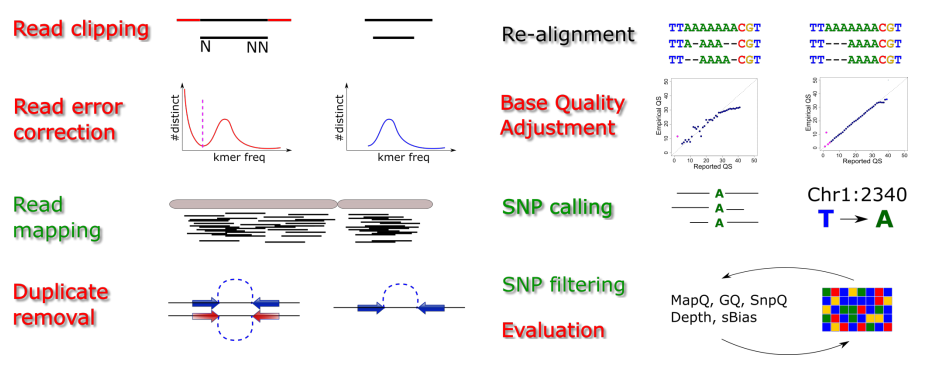 .footnote[ [1][Hands-on Tutorial on SNP Calling](http://www.transplantdb.eu/sites/transplantdb.eu/files/SNPcallingTutorial-Haberer_010715.pdf) ] --- # Somatic short variant discovery (SNVs + Indels) .center[ <img src="./2020-10-05-A_beginners_guide_to_Call_somatic_mutation_Part_IV_files/figure/workflow.png" width="500"> ] .footnote[ [1] [Somatic short variant discovery (SNVs + Indels)](https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels-)] --- # Call SNPs and Indels (in our class) - Somatic SNV and Indels in Solid tumor (not include leukemia) -- - Somatic mutation = mutation in tumor tissue but not in normal tissue or blood -- - SNP (single nucleotide polymorphism) -- - SNV (single nucleotide variant) -- - SNA (single-nucleotide alteration) --- # Make a BAM index file ```bash samtools index [-bc] [-m INT] aln.bam|aln.cram [out.index] ``` Index a coordinate-sorted BAM or CRAM file for fast random access. (Note that this does not work with SAM files even if they are bgzip compressed — to index such files, use tabix(1) instead.) This index is needed when region arguments are used to limit samtools view and similar commands to particular regions of interest. If an output filename is given, the index file will be written to out.index. Otherwise, for a CRAM file aln.cram, index file aln.cram.crai will be created; <u>for a BAM file aln.bam, either aln.bam.bai or aln.bam.csi will be created</u>, depending on the index format selected. [1][Samtools](http://www.htslib.org/doc/samtools.html) --- # Mutect2 ```bash gatk --java-options "-Xmx8G" Mutect2 \ -R /home/admin/database/reference/hg19/ucsc.hg19.fasta \ -I ../out/normal_recal.bam \ -I ../out/cancer_recal.bam \ -tumor cancer \ -normal normal \ --germline-resource af-only-gnomad.raw.sites.hg19.vcf.gz \ -L ../in/Illumina.bed \ -O ../out/somatic.vcf.gz ``` --- # FilterMutectCalls ```bash gatk --java-options "-Xmx8G" FilterMutectCalls \ -V ../out/somatic.vcf.gz \ -O ../out/somatic_filtered.vcf.gz ```  .footnote[ [1] [Filter somatic SNVs and indels called by Mutect2](https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.6.0/org_broadinstitute_hellbender_tools_walkers_mutect_FilterMutectCalls.php) ] --- # VCF format: Meta-information lines ``` ##fileformat=VCFv4.0 ##fileDate=20090805 ##source=myImputationProgramV3.1 ##reference=1000GenomesPilot-NCBI36 ##phasing=partial ``` File meta-information is included after the ## string, often as key=value pairs. .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Meta-information lines ``` ##INFO=<ID=NS,Number=1,Type=Integer,Description="NumberOfSamplesWithData"> ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth"> ##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency"> ##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele"> ##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP membership, build 129"> ##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership"> ``` INFO fields should be described as follows (all keys are required): ``` ##INFO=<ID=ID,Number=number,Type=type,Description=”description”> ``` Possible Types for INFO fields are: Integer, Float, Flag, Character, and String. .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Meta-information lines ``` ##FILTER=<ID=q10,Description="Quality below 10"> ##FILTER=<ID=s50,Description="Less than 50% of samples have data"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality"> ``` FILTERs that have been applied to the data should be described as follows: ``` ##FILTER=<ID=ID,Description=”description”> ``` Likewise, Genotype fields specified in the FORMAT field should be described as follows: ``` ##FORMAT=<ID=ID,Number=number,Type=type,Description=”description”> ``` Possible Types for FORMAT fields are: Integer, Float, Character, and String. .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: The header line syntax ``` #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002 NA00003 ``` The header line names the 8 fixed, mandatory columns. These columns are as follows: - CHROM - POS - ID - REF - ALT - QUAL - FILTER - INFO If genotype data is present in the file, these are followed by a FORMAT column header, then an arbitrary number of sample IDs. The header line is tab-delimited. .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines 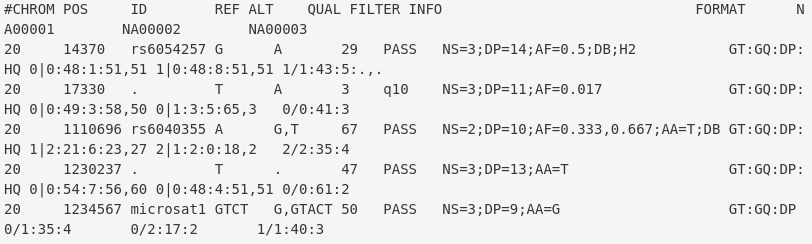 There are 8 fixed fields per record. All data lines are tab-delimited. In all cases, missing values are specified with a dot (”.”). Fixed fields are: 1.CHROM chromosome: an identifier from the reference genome. All entries for a specific CHROM should form a contiguous block within the VCF file.(Alphanumeric String, Required) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  2.POS position: The reference position, with the 1st base having position 1. Positions are sorted numerically, in increasing order, within each reference sequence CHROM. (Integer, Required) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  3.ID semi-colon separated list of unique identifiers where available. If this is a dbSNP variant it is encouraged to use the rs number(s). No identifier should be present in more than one data record. If there is no identifier available, then the missing value should be used. (Alphanumeric String) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  4.REF reference base(s): Each base must be one of A,C,G,T,N. Bases should be in uppercase. Multiple bases are permitted. The value in the POS field refers to the position of the first base in the String. For InDels, the reference String must include the base before the event (which must be reflected in the POS field). (String, Required). .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  5.ALT comma separated list of alternate non-reference alleles called on at least one of the samples. Options are base Strings made up of the bases A,C,G,T,N, or an angle-bracketed ID String (”<ID>“). If there are no alternative alleles, then the missing value should be used. Bases should be in uppercase. (Alphanumeric String; no whitespace, commas, or angle-brackets are permitted in the ID String itself) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  5.QUAL phred-scaled quality score for the assertion made in ALT. i.e. give -10log_10 prob(call in ALT is wrong). If ALT is ”.” (no variant) then this is -10log_10 p(variant), and if ALT is not ”.” this is -10log_10 p(no variant). High QUAL scores indicate high confidence calls. Although traditionally people use integer phred scores, this field is permitted to be a floating point to enable higher resolution for low confidence calls if desired. (Numeric) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  6.FILTER filter: PASS if this position has passed all filters, i.e. a call is made at this position. Otherwise, if the site has not passed all filters, a semicolon-separated list of codes for filters that fail. e.g. “q10;s50” might indicate that at this site the quality is below 10 and the number of samples with data is below 50% of the total number of samples. “0” is reserved and should not be used as a filter String. If filters have not been applied, then this field should be set to the missing value. (Alphanumeric String) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  7.INFO additional information: (Alphanumeric String) INFO fields are encoded as a semicolon-separated series of short keys with optional values in the format: <key>=<data>[,data]. Arbitrary keys are permitted, although the following sub-fields are reserved (albeit optional): - AF allele frequency for each ALT allele in the same order as listed: use this when estimated from primary data, not called genotypes - DP combined depth across samples, e.g. DP=154 .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  ### FORMAT: - 1.GT: genotype. The allele values are 0 for the reference allele (what is in the reference sequence), 1 for the first allele listed in ALT, 2 for the second allele list in ALT and so on. - 2.DP: read depth at this position for this sample (Integer) .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # VCF format: Data lines  - 3.AD: Allelic depths for the ref and alt alleles in theorder listed - 4.AF: Allele fractions of alternate alleles in the tumor .footnote[ [1] [VCF (Variant Call Format) version 4.0](https://www.internationalgenome.org/wiki/Analysis/vcf4.0/) ] --- # Annotation: Annovar ```bash perl /home/admin/software/annovar/table_annovar.pl $INPUT \ /home/admin/software/annovar/humandb -buildver hg19 \ -out $OUT_DIR/$PREFIX.annovar -remove \ -protocol refGene,clinvar_20170905 \ -operation g,f \ -nastring . \ -vcfinput ``` ``` MTOR:NM_004958:exon40:c.C5665G:p.F1888L ``` - gene symbol - transcript id - exon number - cHGVS (info of the variant in coding level) - pHGVS (info of the variant in protein level) --- # Quiz 1.Could you write the commands of call SNVs.Indel and annotation by yourself? Please write your codes in a script file (script.sh). 2.Read the Sequence Variant Nomenclature by yourself. http://varnomen.hgvs.org/